Biography
I am a Statistician and Data Scientist at the Department of Clinical Science, Lund University, Lund, Sweden. My work focuses on developing probabilistic predictive tools that combine computational, experimental, and clinical methods in medical science. I am also researching the structural comparison of proteins and the genetic variations in disease-related biological pathways.
Previously, I was a research fellow in Statistical Machine Learning at André Lab in the Center for Molecular Protein Science, Lund University, where I explored probabilistic models in protein modeling and prediction. Before that, I served as a tenure-track assistant professor of Statistics at the Department of Statistics in Shiraz University and Persian Gulf University between 2015 and 2019.
I have collaborated extensively with Biologists, Medical Physicians, Epidemiologists, Physicists, and Computer Scientists to translate interdisciplinary challenges into computational frameworks. My research interests include statistical machine learning, directional statistics, non-parametric modeling, and Bayesian statistics, with applications in bioinformatics, neuroimaging, and the sustainable development of AI systems to understand complex systems.
This website will take you to my recent research papers and reports, teaching material, external activities, and blog posts. You are welcome to contact me if you are interested in connecting.
Download my resumé.
- Statistical Machine learning
- Directional Statistics
- Bayesian Modeling
- Non-parametric Modeling
PhD in Statistics, 2015
Shahid Beheshti University
Msc in Mathematical Statistics, 2010
Tarbiat Modares University
BSc in Statistics, 2008
Persian Gulf University
Skills
100%
100%
100%
Experience
Responsibilities include:
- Taught Advanced R and C++ Programming, Statistical Modeling and Simulation Studies, Probability, and Linear Algebra courses (70%).
- Researching Statistical Machine Learning and Directional Statistics (30%)
- Member of entrepreneur and innovation committee
Responsibilities include:
- Analysing
- Modelling
- Deploying
- Strategic Planing
- Project Management
Responsibilities include:
- Taught Statistical Bayesian Modeling, Simulation Studies, Probability, Statistical Mehtods, and Linear Algebra courses (70%).
- Researching Statistical Machine Learning and Directional Statistics (30%)
- President’s Management Advisory Board member in Statistics and Strategic Planning
Featured Publications
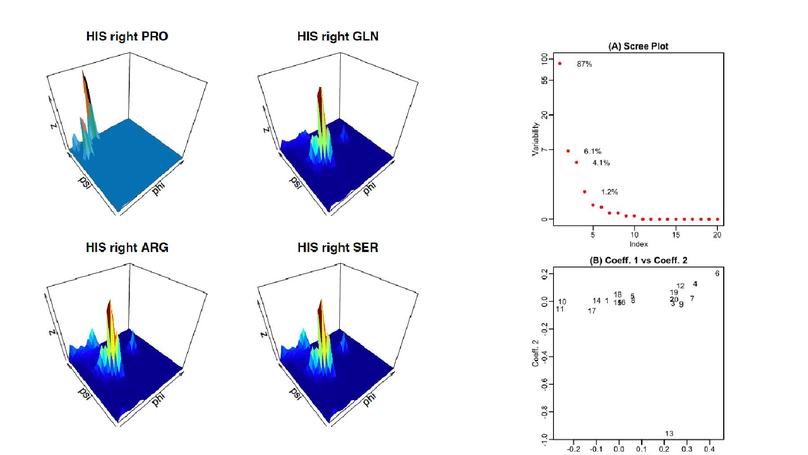
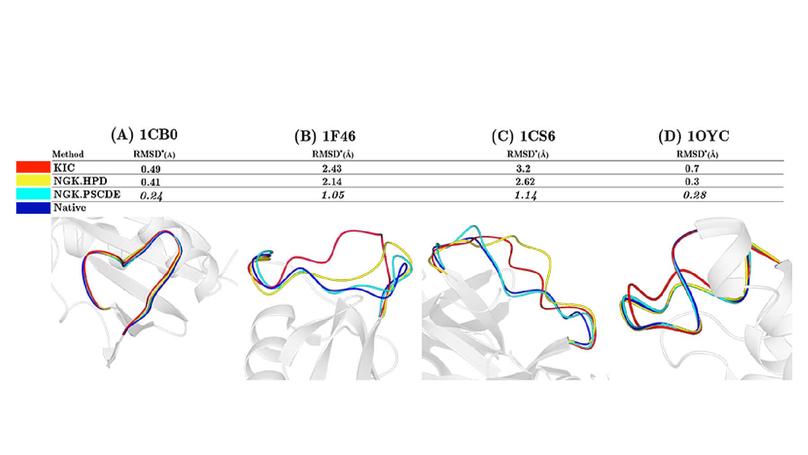
We considered the circular nature of the angular data using trigonometric spline, which was more efficient than the triangulation technique. This general framework also provides comprehensive machinery for clustering, model assessment, or data modeling for groups of protein backbone angles. Specifically, the estimated angular density corresponding to a protein structure has a basis expansion whose coefficients can be used as an input to a clustering algorithm. Furthermore, most of the existing protein classification techniques use sequence and 3D structure comparison to classify the proteins based on some (dis)similarity scores obtained after pairwise alignments. The proposed method is an alignment-free procedure that provides a vector of coefficients (i.e., features) associated with each structure (density) that can be directly used to classify the proteins. This general framework also provides a comprehensive means for assessing clustering models for various other data groups with circular nature. We also developed a shiny web application available at https://pscde-t.shinyapps.io/PSCDE-T/) that can be used by the research community to reproduce the results in this paper and estimate Ramachandran distributions collectively
Contact
- morteza.najibi--at--med.lu.se
- (+46) 76 160 5054
- Kioskgatan 3, SUS, Lund, Scåne 222 42
- DM Me on Twitter








